Study Results
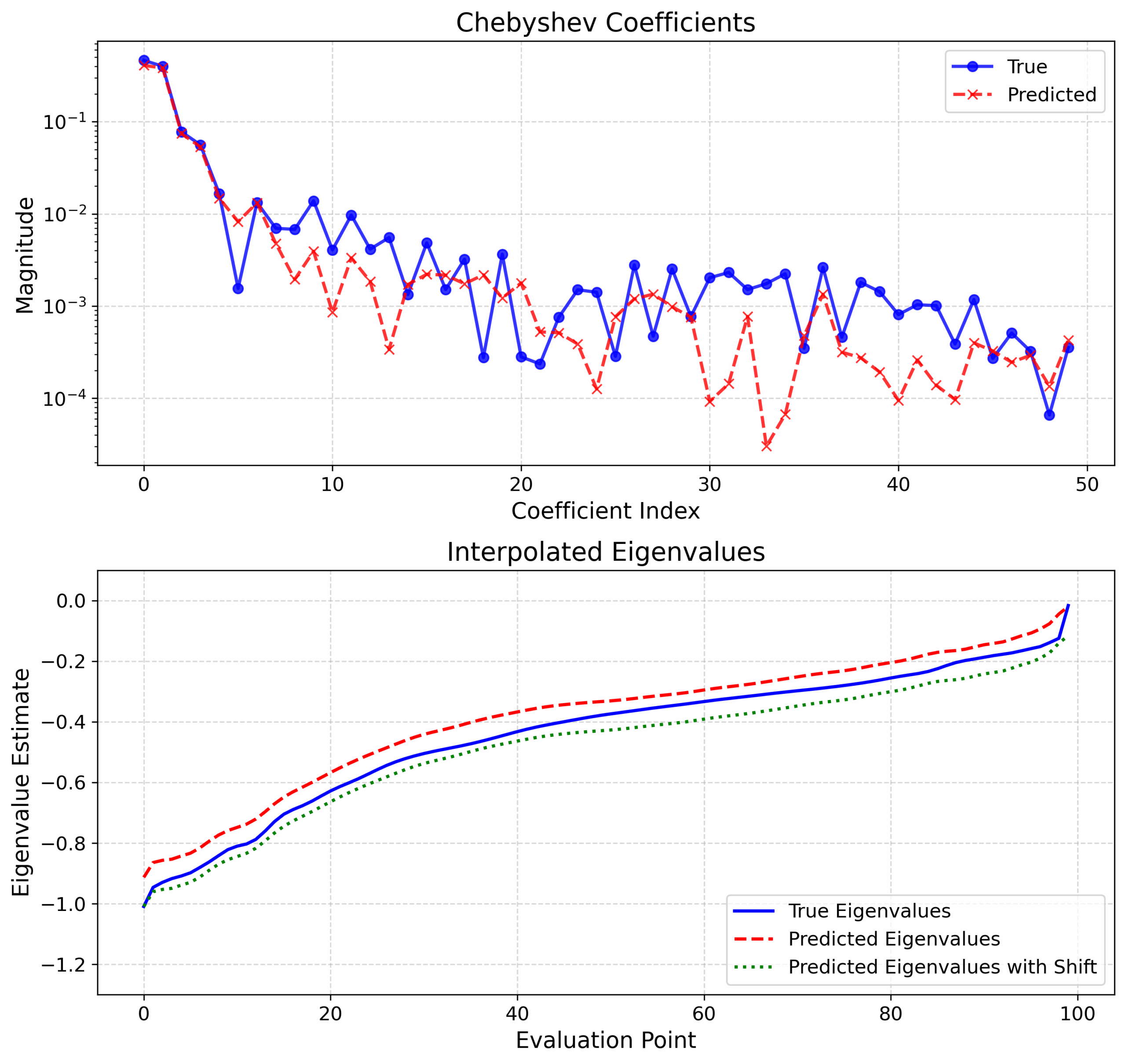
The project successfully applied spectral prediction models to 660 protein dimer systems, generating eigenvalue estimates whose equivalent precision is capable of significantly improving SCF convergence of observables. The predicted density of states (DoS) closely matches the computed reference, demonstrating that the model provides reliable approximations of spectral properties. This approach is in this way capable of effectively reducing the number of SCF iterations required for self-consistency, enhancing the computational efficiency of large-scale quantum mechanical simulations. Albeit preliminary, these results validate the model’s potential for large biomolecular system simulations, paving the way for its integration into automated workflows for protein-ligand interaction studies
Benefits
This study presents multiple benefits across different domains:
- Scientific Advancements: By integrating machine learning with quantum simulations, the project introduces a novel approach to accelerating electronic structure calculations. This will enable large, high throughput biomolecular system simulations that were previously infeasible due to computational constraints.
- Industrial Impact: The ability to rapidly and accurately predict binding interactions in protein-ligand complexes has implications for drug discovery and materials science. This could shorten lead times in pharmaceutical development and improve materials screening processes.
Exascale Computing: The developed model showcases efficient utilization of HPC resources, making it a potential candidate for exascale-ready electronic structure
Partners
| Forschungszentrum Juelich is the largest Helmoltz center in the Germany and hosts the Juelich Supercomputing Center (JSC). The Simulation and Data Lab Quantum Materials (SDLQM) is part of the JSC and provides HPC-expertise as well as the connection between Quantum Materials simulations and simulating clusters. In this project, SDLQM hosted the data and provided the compute time to train the statistical models (KKR) and solve the eigenvalue problems (SLEPc, FRASE). |
| Ruđer Bošković Institute (RBI) is the largest Croatian research institute with the main research focus on natural science. RBI had the role of HPC expert and software developer working on input data (matrices) pre-processing on HPC systems and development of two spectral predictors based on Graph Neural Networks (GNN) and Radial distribution function (RDF)-based models with Random Forest (RF). |
| The French Alternative Energies and Atomic Energy Commission (CEA) is a leading research organization in nuclear energy, AI, and HPC. BigDFT is a wavelet-based Density Functional Theory (DFT) code designed for scalable electronic structure calculations, particularly on large-scale HPC architectures. The BigDFT team generated the data set from the simulations of complex material systems and is the recipient of the statistical algorithms devised. |
Team
- Edoardo Di Napoli
- Xinzhe Wu
- Gustavo Ramirez-Hidalgo
- Davor Davidovic
- Abhiram Kaushik Badrinarayanan
- Jurica Novak
- Luigi Genovese
Contact
Name: Edoardo Di Napoli
Institution: Forschungszentrum Juelich
Email Address: e.di.napoli@fz-juelich.de